
In order for our scaffold to effectively bring together the five T3SS needle tip proteins in close enough proximity for them to self-assemble we need a method of chemically attaching each of the proteins to the DNA scaffold. The method we chose was the Ni-NTA / His-tag interaction, which utilises the strong affinity between NTA modified DNA, Ni2+ ions and hexahistidine tagged protein. Conjugation DNA was produced by modifying tri-aminated DNA with NTA groups [1] and then purifying the tris-NTA DNA away from the partially converted forms.
To modify tri-aminated DNA by the addition of three NTA groups.
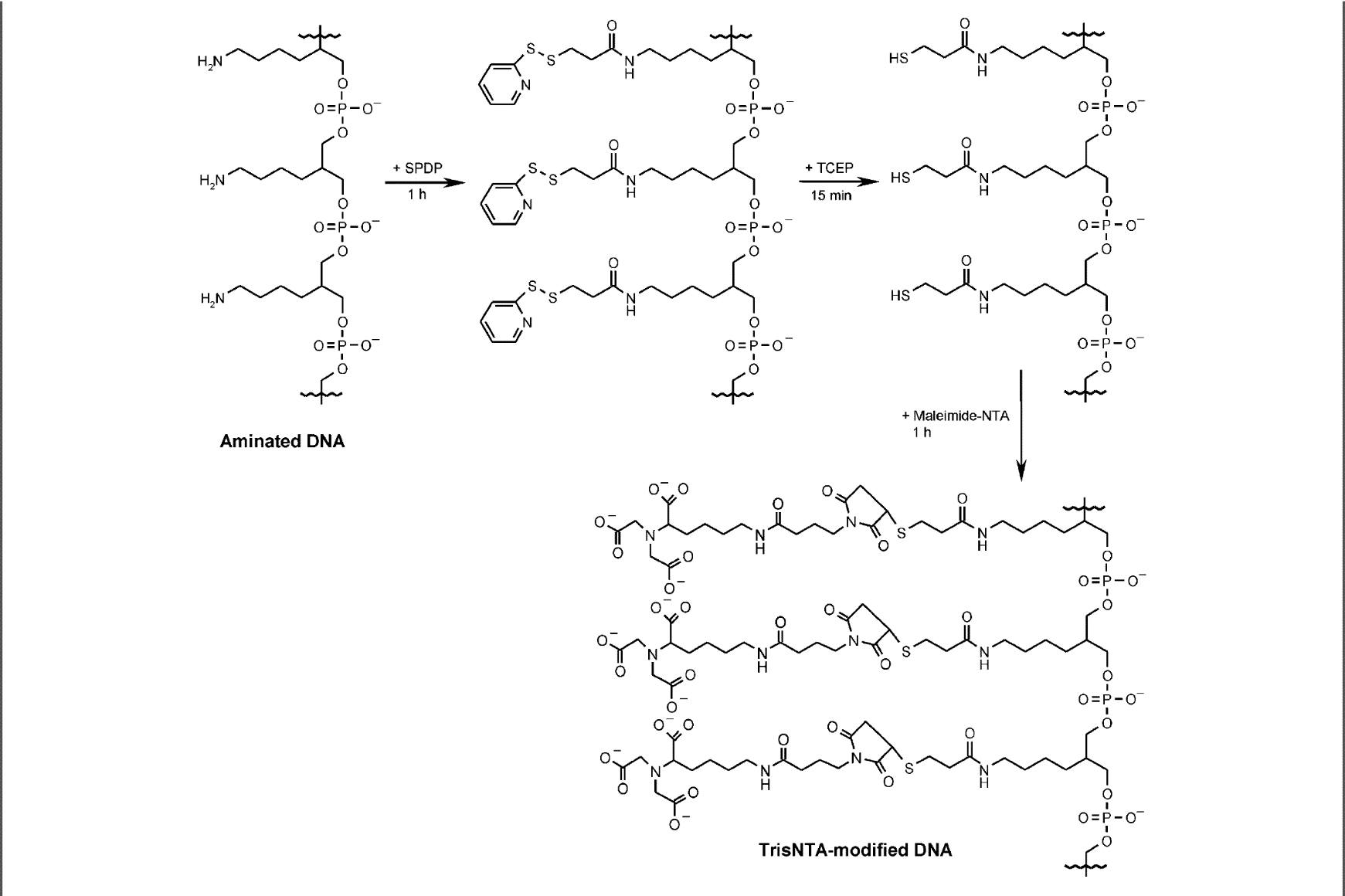
Figure 1. Reaction schematic of the NTA modification process [1]
Tri-aminated DNA was resuspended in nuclease-free water to 1 mM. 50 uL of DNA was buffer exchanged into phosphate buffer using a pre-equilibrated SEC spin column (Bio-Rad Micro Bio-Spin 6 - exclusion limit 5 bp or 6 kDa proteins/peptides). 12.5 uL of SPDP was added to the DNA solution and left for 1 hour at room temperature. Excess SPDP was removed using SEC spin columns (use 2 columns per 50 uL DNA solution). 6.25 uL of 100 mM TCEP was added to the DNA solution and left for 15 min at room temperature. 7 uL of 200 mM maleimide-NTA was added and left for 1 hour at room temperature. Excess maleimide-NTA was removed with SEC spin columns pre-equilibrated in Phosphate Buffer. DNA can be stored at 4 oC for several days, but best to purify it as soon as possible.
All steps of the modification process were visualised using denaturing PAGE gel. These confirmed tris-NTA DNA was successfully produced as the major product, with significant amounts of bis-, mono-NTA and unmodified DNA also produced as by-products due to incomplete reaction (Figure 2). Thus, the crude product required purification to isolate pure tris-NTA DNA.
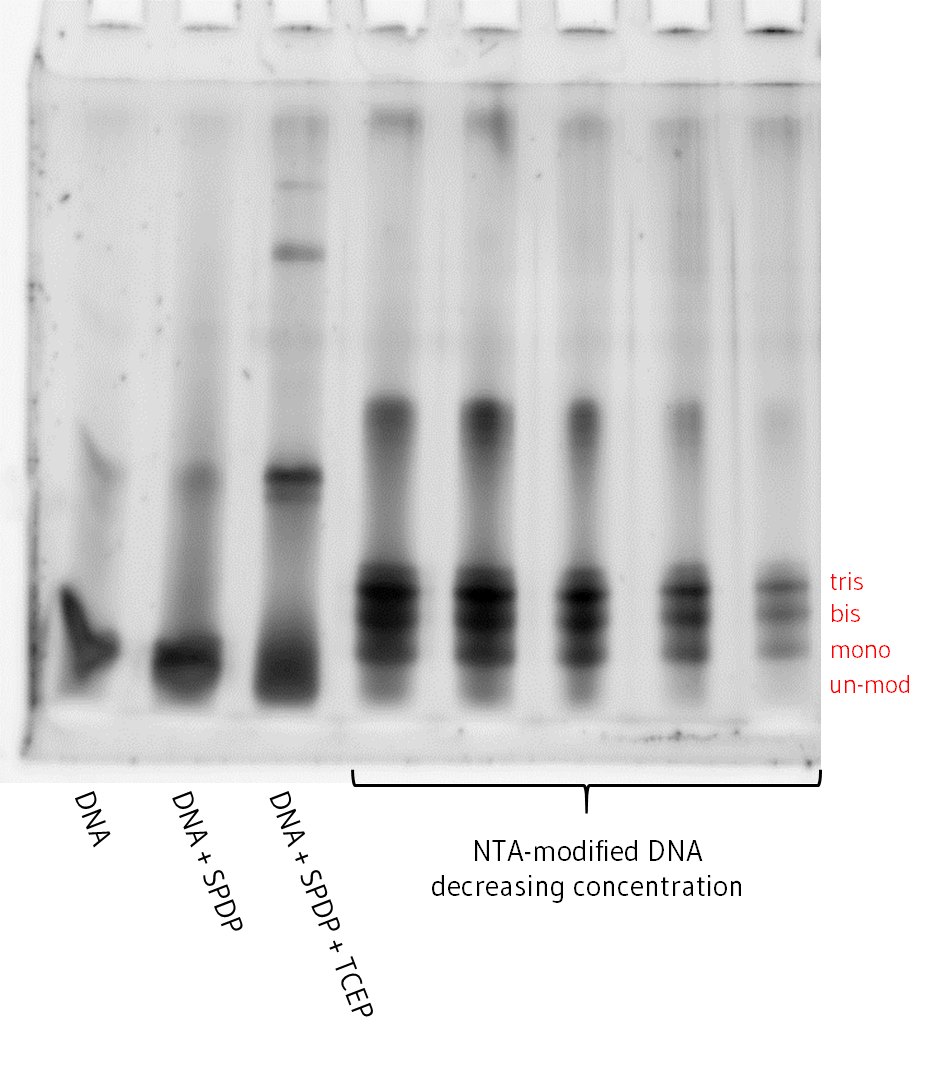
Figure 2. Denaturing PAGE gel showing samples at each stage of the chemical modification process
To isolate and purify tris-NTA modified DNA away from bis-, mono-NTA DNA and other impurities.
Tris-NTA modified DNA was purified by gel extraction. Sample was prepared by combining 10 uL of modified DNA with 190 uL of MilliQ water and 200 uL of 2x urea loading dye (total volume 400uL). 12.5 uL was loaded into each of the middle 8 wells of 4 x 10-well denaturing PAGE gels. Gel electrophoresis was run at room temperature for 10 minutes at 150V, then 90 minutes at 200V to separate partially modified DNA from Tris-NTA DNA. The gels were stained in 50mL Tris-Glycine and 5uL SYBR Gold for 10 minutes in a shaker. This showed 4 clear bands corresponding to unmodified, mono, bis and tris modified DNA (Figure 3).
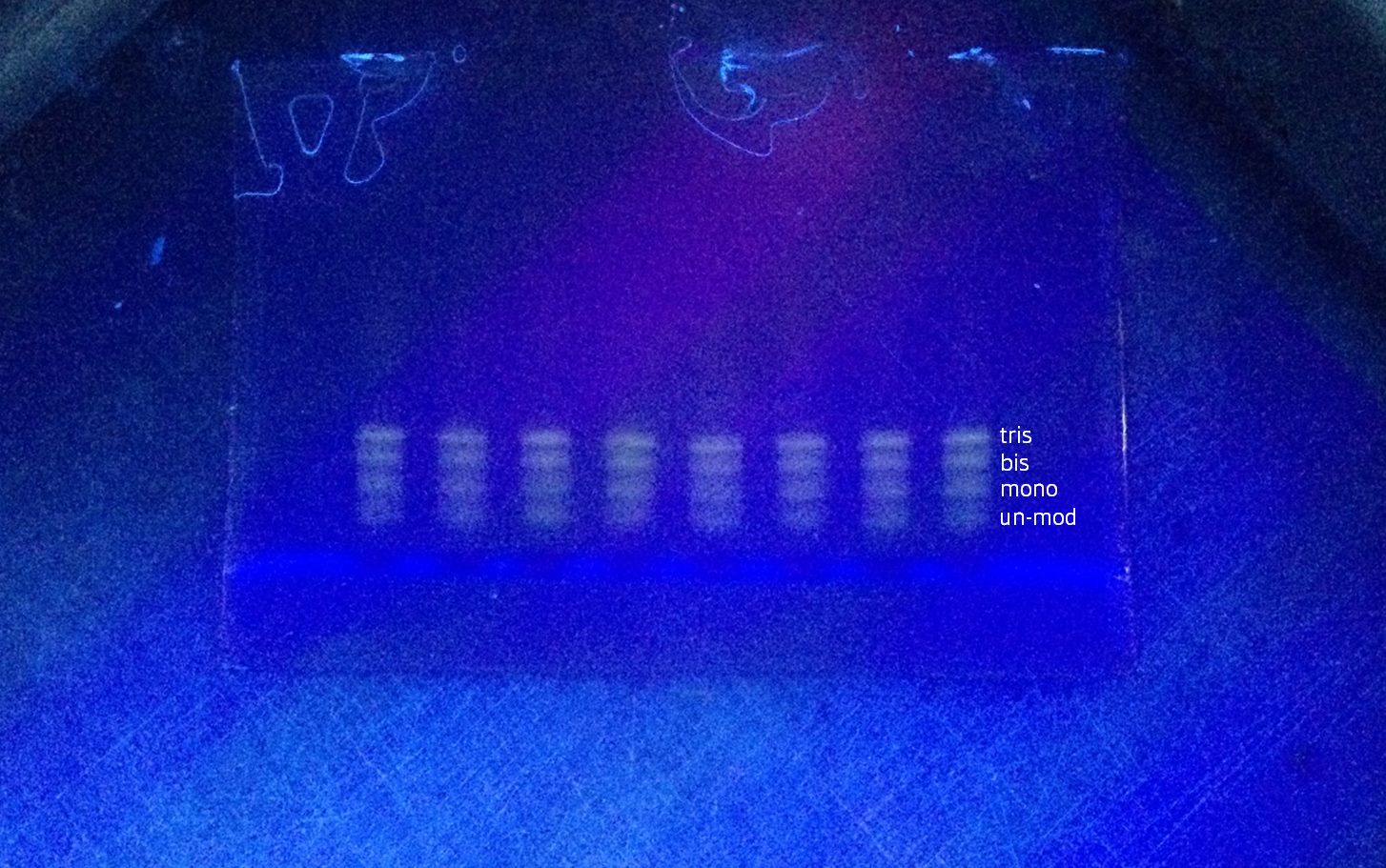
Figure 3. Denaturing PAGE gel on UV light box before removing top tris-NTA DNA band; showing tris, bis, mono-NTA and unmodified bands
Purified tris-NTA DNA was extracted by cutting a strip of the gel corresponding to the highest major band under UV light. To redissolve the gel, this was transferred to a separate ‘crushing tube’ consisting of a 500 uL eppendorf tube with a hole poked through the bottom with a red hot needle placed inside a larger 2 mL eppendorf tube. The tubes were centrifuged at 12500g for 5 minutes, which crushed the gel by extruding it through a small orifice. 300 uL of 20mM Tris HCl pH 8 was added to resuspend the crushed gel and left to incubate on a horizontal roller overnight.
Dissolved Tris-NTA DNA was filtered from the gel using 0.45 um cellulose acetate SEC spin filter columns and centrifuged at 12500g for 5 minutes. The filtrates were pooled (sample #1). A second extraction from the gel was performed using 200 uL of 20mM Tris HCl pH 8 and (sample #2). Samples #1 and #2 were concentrated using speedy vac for ~90 mins until the volume was below 75 uL. Samples were buffer exchanged into filtered MilliQ water using G10 resin buffer exchange spin columns. At each stage of the purification, 7 ul of sample was set aside for analysis with PAGE to assess purity (Figure 4), which was then quantified by measuring absorbance at 280 nm (Table 1).
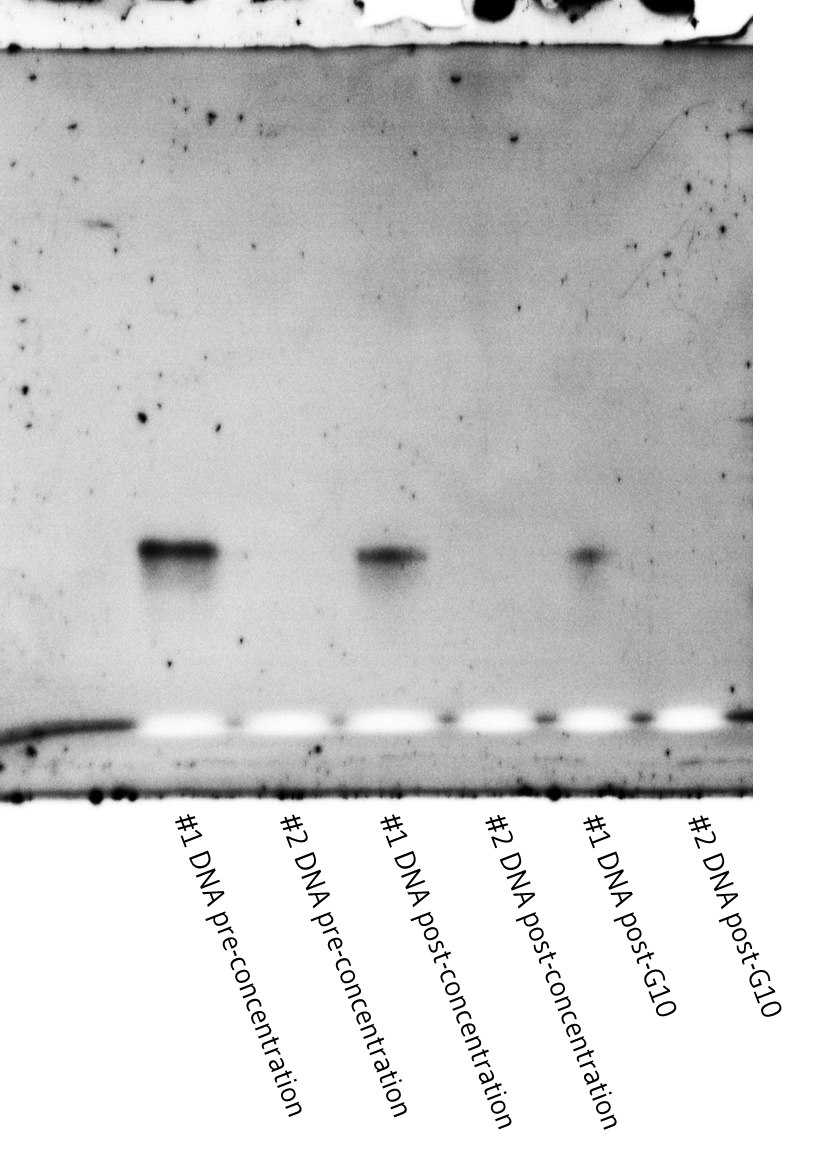
Figure 4. Denaturing PAGE gel showing stages of the purification process for samples #1 and #2.
Table 1. Yield data for each run of experiment 2
| DNA loaded on gels | Pre-conc. (ng) | Post-conc. (ng) | Post-G10 (ng) | Yield |
| 1. 10 nt 5' sample #1 | 7395 | 4774 | 3441 | 46% |
| 1. 10 nt 5' sample #2 | 3021 | 2496 | 1746 | 47% |
| 2. 20 nt 3' sample #1 | 12798 | 7770 | 5925 | 46% |
| 2. 20 nt 3' sample #2 | 3690 | 3735 | 2106 | 57% |
| 3. 20 nt 3' sample #1 | 3150 | 2125 | 1512 | 48% |
| 3. 20 nt 3' sample #2 | 1386 | 659 | 559 | 40% |
| 4. 20 nt 3' sample #1 | 2880 | 2849 | 2273 | 79% |
| 4. 20 nt 3' sample #2 | 913 | 800 | 616 | 68% |
| 5. 20 nt 3' sample #1 | 4288 | 6570 | 2226 | 52% |
| 5. 20 nt 3' sample #2 | 1060 | 840 | 312 | 29% |
PAGE gels (Figure 4) confirmed the purity of the batches of tris-NTA DNA isolated, indicating that the purifications were successful. The gels however showed little to no DNA in sample #2 in each case, despite the nano-drop consistently measuring non-negligible concentrations. The lack of DNA in the second extraction suggests that most of the DNA was successfully extracted in the first extraction. The positive concentration readings on the nano-drop are then possibly due to elements of the gel becoming saturated in the Tris buffer in the extraction step and flowing through the 0.45 um SEC spin column.
We successfully modified and purified both 10 nt and 20 nt tris-NTA DNA with base sequences complimentary to the binding site strands in our designed scaffolds. These NTA-modified strands were crucial to the rest of the project as they gave us the chemical method of reliably attaching hexahistidine tagged proteins to our DNA scaffolds.
10 nt 5' tri-aminated DNA
Sequence: 5' GGCAGGACGG 3'
20 nt 3' tri aminated DNA
Sequence: 5' ACTATCGCATCAACAGGACA 3'
Phosphate buffer (100mM sodium phosphate, 100mM NaCl, pH 7.3)
75mM SPDP (in DMSO)
100mM TCEP
200mM Maleimide-NTA
| 1x Resolving Gel | 4x Resolving Gel | 1x Stacking Gel | 4x Stacking Gel | |
| Urea (g) | 2.1 | 8.4 | 0.84 | 3.36 |
| 3 M Tris-HCl, pH 8.8 (mL) | 0.65 | 2.6 | - | - |
| 1 M Tris-HCl, pH 6.8 (mL) | - | - | 0.25 | 1 |
| 40% acrylamide/bis-acrylamide (mL) | 2.75 | 11 | - | - |
| 30% acrylamide/bis-acrylamide (mL) | - | - | 0.33 | 1.32 |
| MilliQ water (mL) | - | - | 1 | 4 |
| 10% APS (uL) | 25 | 100 | 10 | 40 |
| TEMED (uL) | 5 | 25 | 2.5 | 10 |
0.42 g Urea
93.75 uL 1 M Tris-HCl, pH 6.8
62.5 uL 1% bromophenol blue
25mM Tris
0.19M Glycine
20mM Tris-HCl, pH 8.0
1. Goodman, R. P. et al. A facile method for reversibly linking a recombinant protein to DNA. ChemBioChem 10, 1551-1557 (2009).